Usage
This section details usage of GRAViTy-V2 from the GUI only, and assumes that you are not running GRAViTy-V2 from a Docker container. For CLI and container guidance, see Advanced usage options.
To run GRAViTy-V2, users need to provide a vmr-like .csv document with details of viruses to be analysed. Optionally, users can also provide input sequences in GenBank format for cases where sequences are not able to be scraped automatically from GenBank. This file is passed to GRAViTy-V2 via the "GenomeDescTableFile" parameter.
GRAViTy-V2 jobs are run on reference sequences (downloaded from GenBank), user-provided sequences or a combination of these. Users must specify a file path for a GenBank file via the "GenomeSeqFile" parameter. If this file doesn't exist, GRAViTy-V2 will attempt to download all sequences from GenBank and save them to this file, using the Accession ID field in the input VMR. If you give GRAViTy-V2 a ready-made GenBank file, it will attempt to use this; if it doesn't contain all of the sequences you specified in the VMR, it will attempt to download the missing sequences from GenBank.
GRAViTy-V2 is packaged with several utility functions to help you generate VMR-like documents, scrape the latest VMR from the ICTV, convert multi-FASTA files to GenBank format and combine reference and user-provided sequence files.
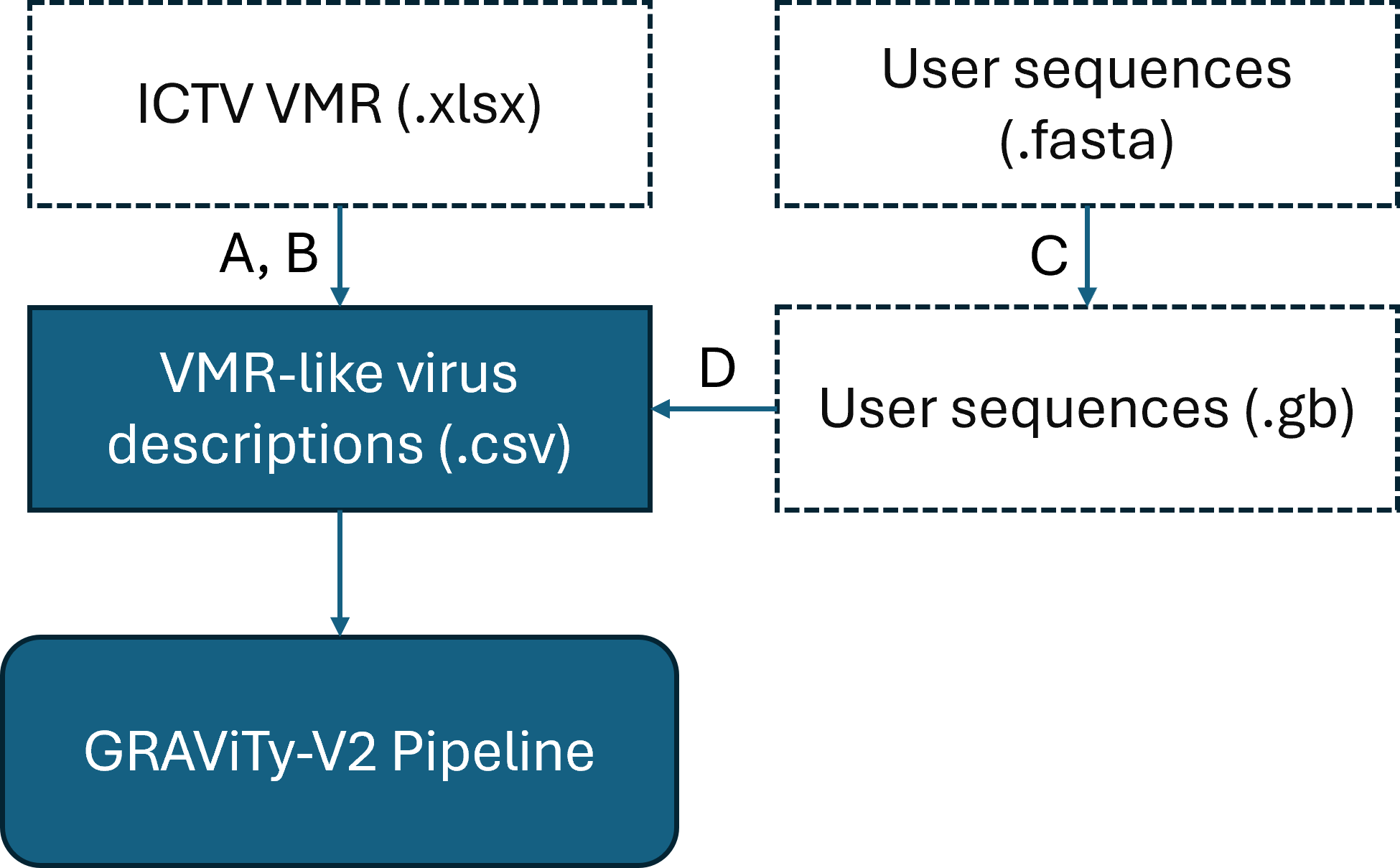
GRAViTy-V2 Input Data & Utility Functions
Dashed line boxes are optional. A: Scrape VMR; B: Filter VMR; C: Convert FASTA to GenBank; D: Concatenate user sequences to VMR
The remainder of this section follows the use of all GRAViTy-V2 utility functions to create an example workflow in which we generate a new classification from scratch. We start with a single sequence (.fasta format, included in this repository) that we know is a member of Hepacivirus, and build a workflow to classify it against other classified members of this genus.
The ICTV's virus metadata resource (VMR) is a tabular file containing details of all of the currently classified taxa, which is used by GRAViTy-V2 as a template for metadata and downloading reference sequences. This function will download the latest VMR and structure it ready to be filtered for use in your GRAViTy-V2 experiments.
- Start and open GRAViTy-V2's API
- Find the
scrape_vmrendpoint. Expand the dialogue box. - Click "try it now". (If required, users can here change the output file name (what you want your VMR to be called) and save directory.)
- Click "execute"
Two filter functions exists to take your newly-downloaded VMR and refine it to a smaller list of viruses to use as reference sequences in your classification. This example shows use of the first pass filter function to filter the VMR we just scraped to only Hepacivirus sequences.
- Start and open GRAViTy-V2's GUI, and follow the instructions above for scraping a VMR.
- Find the
filter_vmr_first_passendpoint, expand the box and click "try it now" - Paste the following code box into the dialogue box. This will filter the scraped VMR as follows: maximum of 100 sequences will be selected (randomly; "filter_threshold"), to include only viruses who are labelled "Hepacivirus" in the "Genus" column of the VMR ("filter_name", "filter_level"). The file it filters will be "data/latest_vmr.csv" ("vmr_name") and it will save the filtered vmr-like document to "data/latest_vmr_hepacivirus_filter.csv" ("save_path"/"save_name")
{
"filter_threshold": 100,
"save_name": "latest_vmr_hepacivirus_filter.csv",
"filter_level": "Genus",
"filter_name": "Hepacivirus",
"vmr_name": "data/latest_vmr.csv",
"save_path": "./data/"
}
We will now convert our hypothetical unclassified sequence (which is included in the repository) from FASTA to GenBank, so that it can be used as input to the GRAViTy-V2 pipeline. This function also generates a VMR-like document, which will later be combined with the filtered VMR we created in the previous step
- Start and open GRAViTy-V2's GUI, and follow steps 1-2 above.
- Find the
convert_fasta_to_genbank_and_vmrendpoint, expand the box and click "try it now". - Paste the following code box into the dialogue box. This will read the fasta file ("fasta_fname") and save corresponding genbank ("genbank_fname") and vmr-like document ("vmr_fname") files.
{
"fasta_fname": "data/eval/example_seq.fasta",
"genbank_fname": "data/example_seq.gb",
"vmr_fname": "data/example_seq.csv"
}
We will now join the VMR-like documents created in steps 2 and 3, such that a GRAViTy-V2 experiment can be run that compares the unclassified sequence against all of the species within Hepacivirus.
- Start and open GRAViTy-V2's GUI, and follow steps 1-3 above.
- Find the
join_vmrsendpoint, expand the box and click "try it now". - Paste the following code box into the dialogue box. This joins our two documents ("vmr_1", "vmr_2") and saves the output to a new file ("vmr_joined")
{
"vmr_1": "./data/latest_vmr_hepacivirus_filter.csv",
"vmr_2": "./data/example_seq.csv",
"vmr_joined": "./data/GRAViTy-V2_hepacivirus_test.csv"
}
We will now run our GRAViTy-V2 experiment, using one of the premade pipelines (details of different workflows may be found in consequent sections, below), which is optimised for use with datasets containing all similar viruses. N.b. we only provide one unclassified sequence in the input GenBank file; GRAViTy-V2 will detect that all of the Hepacivirus sequences are missing and download them from GenBank.
- Start and open GRAViTy-V2's GUI, and follow steps 1-4 above.
- Find the
similar_virusesendpoint, expand the box and click "try it now". - Paste the following code box into the dialogue box. This runs the premade similar viruses GRAViTy-V2 pipeline, using the joined VMR-like document of sequence descriptions ("GenomeDescTableFile") and GenBank file containing the unclassified sequence ("GenomeSeqFile"). Output data are saved to a folder called ./output/GRAViTy-V2_hepacivirus_test ("ExpDir").
{
"GenomeDescTableFile": "./data/GRAViTy-V2_hepacivirus_test.csv",
"ExpDir": "./output/GRAViTy-V2_hepacivirus_test",
"GenomeSeqFile": "./data/example_seq.gb"
}
Interpretation of this experiment's output is included in Output file interpretation.
There are two types of analysis that GRAViTy-V2 can perform:
- Creating a new taxonomic classification
- Appending new virus genomes to an existing classification from (1)
For (1), users may opt for either a premade workflow, in which we've created several easy-to-use pipeline entrypoints, or an new classification pipeline over which users may have full control. For (2), we don't offer premade workflows, so users must make define a custom end-to-end job using the update classsification pipeline.
Premade pipelines are adapted for common use cases --- datasets comprising similar viruses, divergent viruses and viruses with long, single ORFs --- and require only minimal user input to run. See the guided example above for an example of how to use the premade pipelines.
The new_classification endpoints allow users full control of GRAViTy-V2 parameters. Details of how to run an equivalent experiment to the one shown in the guided experiment above are found below, and assumes you have the input files from this. Descriptions of these parameters are found in another section, below.
There are 5 new classification endpoints. Most users will need the new_classification_full endpoint as this runs a complete, end-to-end pipeline job. The other endpoints start a new classification job part-way through the pipeline and are useful when users want to change parameters without having to re-run the entire pipeline. E.g. if users want to re-generate graphs with a different bootstrap support cutoff ("Heatmap_DendrogramSupport_Cutoff"), they can run the new_classification_from_graph_generator endpoint. If users attempt to start the pipeline mid-way through without having previously run the initial stages, GRAViTy-V2 will produce an error.
- Start and open GRAViTy-V2's GUI, and follow steps 1-4 in the guided example, above.
- Find the
new_classification_fullendpoint, expand the box and click "try it now". - Paste the following code box into the dialogue box.
{
"GenomeDescTableFile": "data/GRAViTy-V2_hepacivirus_test.csv",
"ExpDir": "./output/GRAViTy-V2_hepacivirus_test",
"GenomeSeqFile": "./data/example_seq.gb",
"AnnotateIncompleteGenomes": false,
"TaxoGrouping_Header": "Genus",
"genbank_email": "name@provider.com",
"ProteinLength_Cutoff": 100,
"IncludeProteinsFromIncompleteGenomes": true,
"Mash_p_val_cutoff": 0.05,
"Mash_sim_score_cutoff": 0.95,
"ProtClustering_MCLInflation": 2,
"N_AlignmentMerging": 0,
"PPHMMClustering_MCLInflation_ForAlnMerging": 5,
"HMMER_PPHMMDB_ForEachRoundOfPPHMMMerging": true,
"HHsuite_evalue_Cutoff": 1e-06,
"HHsuite_pvalue_Cutoff": 0.05,
"HHsuite_QueryCoverage_Cutoff": 85,
"HHsuite_SubjectCoverage_Cutoff": 85,
"HMMER_C_EValue_Cutoff": 0.001,
"HMMER_HitScore_Cutoff": 0,
"p": 1,
"Dendrogram_LinkageMethod": "average",
"N_Bootstrap": 10,
"Bootstrap_method": "sumtrees",
"VirusGrouping": true,
"SimilarityMeasurementScheme": "PG",
"Heatmap_DendrogramSupport_Cutoff": 0.75,
"PphmmNeighbourhoodWeight": 0,
"PphmmSigScoreThreshold": 0,
"RemoveSingletonPPHMMs": true,
"N_VirusesOfTheClassToIgnore": 1,
"PPHMMSorting": false,
"PPHMMClustering_MCLInflation_ForPPHMMSorting": 2,
"N_Sampling": 10,
"SamplingStrategy": "balance_with_repeat",
"SampleSizePerGroup": 10,
"UseBlast": true,
"NThreads": "auto",
"ClustAlnScheme": "local",
"MutualInformationScorer": false,
"Bootstrap": true,
"N_CPUs": "auto"
}
The update_classification pipeline exists for when you have a stable, ready-made GRAViTy-V2 classification and want to add new sequences to it. There are two reasons users might want to do this:
- This is usually quicker than re-running an entirely new classification
- The original classification remains stable and is not-recalculated, so is useful when comparing two clades with very different characteristics that necessitate the use of different GRAViTy-V2 settings.
To demonstrate such a use case, we will build on the dataset generated in the guided example, above, and assumes you have both the data and the GRAViTy-V2 results generated from it.
- Firstly, we will generate a new VMR-like document with new sequences that we want to compare with our existing classification. We will choose the Pegivirus genus. Input the following code into the
filter_vmr_first_passendpoint.
{
"filter_threshold": 100,
"save_name": "latest_vmr_pegivirus_filter.csv",
"filter_level": "Genus",
"filter_name": "Pegivirus",
"vmr_name": "data/latest_vmr.csv",
"save_path": "./data/"
}
- Find the
update_classification_fullendpoint, expand the box and click "try it now". - Paste the following code box into the dialogue box. "GenomeDescTableFile", "ExpDir_Pl1" and "GenomeSeqFile_RefVirus" are all derived from your original GRAViTy-V2 experiment (see above). For the update classification run, we need to specify a new experiment directory ("ShelveDir_UcfVirus"), a new genbank file to download sequences to ("GenomeSeqFile_UcfVirus") and your newly-filtered VMR ("GenomeDescTableFile_UcfVirus").
{
"GenomeDescTableFile": "data/GRAViTy-V2_hepacivirus_test.csv",
"ExpDir_Pl1": "output/GRAViTy-V2_hepacivirus_test",
"GenomeSeqFiles_RefVirus": "./data/example_seq.gb",
"ShelveDir_UcfVirus": "output/GRAViTy-V2_update_classification_test",
"GenomeSeqFile_UcfVirus": "data/pegivirus_seqs.gb",
"GenomeDescTableFile_UcfVirus": "data/latest_vmr_pegivirus_filter.csv",
"AnnotateIncompleteGenomes": false,
"TaxoGrouping_Header": "Genus",
"genbank_email": "name@provider.com",
"ProteinLength_Cutoff": 100,
"IncludeProteinsFromIncompleteGenomes": true,
"Mash_p_val_cutoff": 0.05,
"Mash_sim_score_cutoff": 0.95,
"ProtClustering_MCLInflation": 2,
"N_AlignmentMerging": 0,
"PPHMMClustering_MCLInflation_ForAlnMerging": 5,
"HMMER_PPHMMDB_ForEachRoundOfPPHMMMerging": true,
"HHsuite_evalue_Cutoff": 0.000001,
"HHsuite_pvalue_Cutoff": 0.05,
"HHsuite_QueryCoverage_Cutoff": 85,
"HHsuite_SubjectCoverage_Cutoff": 85,
"HMMER_C_EValue_Cutoff": 0.001,
"HMMER_HitScore_Cutoff": 0,
"p": 1,
"Dendrogram_LinkageMethod": "average",
"N_Bootstrap": 10,
"Bootstrap_method": "sumtrees",
"VirusGrouping": true,
"SimilarityMeasurementScheme": "PG",
"Heatmap_DendrogramSupport_Cutoff": 0.75,
"PphmmNeighbourhoodWeight": 0.0125,
"PphmmSigScoreThreshold": 0,
"UseBlast": false,
"NThreads": "auto",
"ClustAlnScheme": "local",
"UseUcfVirusPPHMMs": true,
"AnnotateIncompleteGenomes_UcfVirus": true,
"UsingDatabaseIncludingIncompleteRefViruses": false,
"DatabaseAssignmentSimilarityScore_Cutoff": 0.01,
"N_PairwiseSimilarityScores": 10000
}
Output file interpretation may be found in Output file interpretation.
- GenomeDescTableFile: Path to VMR-like document (CSV)
- ExpDir: Directory to save output data. If doesn't exist, GRAViTy-V2 will create.
- GenomeSeqFile: Path to GenBank file containing sequence data. If doesn't exist, GRAViTy-V2 will attempt to download from NCBI Entrez, using virus accession IDs from input VMR (GenomeDescTableFile)
- TaxoGrouping_Header: Column title in VMR to draw taxon boundaries around in output. Choose from "Family", "Genus" "Taxonomic grouping", where latter is a custom column that users can create in the input CSV document.
- genbank_email: Provide an email address for use in the NCBI Entrez sequence download system.
- ProteinLength_Cutoff: Miminum length (in AA) for a protein sequence to be used in GRAViTy-V2 analysis. Recommended range: 70-100.
- Mash_p_val_cutoff: p value above which Mash hit will be ignored (as part of initial ORF clustering, during PPHMMDb construction). Recommended to not change.
- Mash_sim_score_cutoff: similarity score below which Mash hit will be ignored (as part of initial ORF clustering, during PPHMMDb construction). Recommended range 0.7-0.95.
- ProtClustering_MCLInflation: mcl algorithm inflation score (as part of initial ORF clustering, during PPHMMDb construction). Recommended range 2.0-4.0.
- N_AlignmentMerging: Number of iterations for alignment merging rounds (as part of initial ORF clustering, during PPHMMDb construction). Warning: this can dramatically increase run times and has only a few, highly specific applications. Recommended to not change.
- PPHMMClustering_MCLInflation_ForAlnMerging: mcl algorithm inflation score (as part of alignment merging). Argument is ignored if N_AlignmentMerging == 0. Recommended to not change.
- HHsuite_evalue_Cutoff: [IF SORTING PPHMMS]
- HHsuite_pvalue_Cutoff:
- HHsuite_QueryCoverage_Cutoff:
- HHsuite_SubjectCoverage_Cutoff:
- HMMER_C_EValue_Cutoff: Hmmer E value above which to ignore hits (as part of PPHMM signature generator). Recommended to not change.
- HMMER_HitScore_Cutoff: Hmmer hitscore below which to ignore hits (as part of PPHMM signature generator). Recommended range 0-40 (use will be contextual; 25 is a good starting point, although 0 is default).
- p: Scale factor for similarity matrix. Recommended to not change.
- Dendrogram_LinkageMethod: Select from ["single", "complete", "average", "weighted", "centroid", "median", "ward"]
- N_Bootstrap: If > 0, calculate bootstrap support for dendrograms with n iterations (recommended 10 or 0).
- Bootstrap_method: Method for calculating bootstrap support. Select from ["sumtrees", "booster"]
- VirusGrouping: Perform virus grouping (as part of virus classification). Recommended true.
- SimilarityMeasurementScheme: Scheme for constructing similarity matrix. Choose from: ["P", "G", "L", "PG", "PL", "R", "RG", "PR"], based on whether PPHMM signatures (P), GOM signatures (G), distance correlation (L), PPHMM ratios (R) or combinations of these are used. Recommended "PG".
- Heatmap_DendrogramSupport_Cutoff: Threshold for bootstrap support to be shown on GRAViTY-V2 heatmap dendrograms.
- PphmmNeighbourhoodWeight: Apply weighting to profile scores when all genomes being analysed are highly similar (as part of similarity matrix construction). Recommended to not change.
- PphmmSigScoreThreshold: Disregard protein profiles where signature scores are below (as part of similarity matrix construction). Recommended to not change.
- UseBlast: If true, use BLASTp instead of Mash (as part of initial ORF clustering, during PPHMMDb construction). Recommended when comparing datasets comprising mostly very similar viruses (e.g. all within a single family).
- NThreads: Number of threads for multiprocessing. Choose from either an integer or "auto". Recommended to not change, unless users experience out of memory errors or are working on HPCs.
- ClustAlnScheme: Determines which algorithm is used to do protein alignments (as part of initial ORF clustering, during PPHMMDb construction). Choose from ["local", "global", "auto"], to correspond to Mafft L-INS-i, G-INS-i and auto algorithms, respectively. Recommended to "auto" if unsure, as this is the fastest option.
- RemoveSingletonPPHMMs: If true, disregard scores from PPHMMs shared by only n genomes (see N_VirusesOfTheClassToIgnore). Recommended to not change.
- N_VirusesOfTheClassToIgnore: If true and RemoveSingletonPPHMMs true, number of genomes above which must possess a PPHMM for it to be included in the analysis. Recommended to not change.
- PPHMMSorting: If true, sort PPHMMs (as part of PPHMMDb construction). This can dramatically increase run times and will rarely increase performance. Recommended false.
- N_Sampling: Number of mutual information scores sample size. Recommended to not change.
- SamplingStrategy: If N_Sampling > 0, strategy for calculating mutual information.
- SampleSizePerGroup: If N_Sampling > 0, sample size for each group.
- MutualInformationScoring: If true, calculate mutual information score to quantify feature importance of each PPHMM in a run. Useful statistic but may greatly increase run times.