-1.png){kind=link}

The SARS-CoV-2 pandemic has affected many individuals and completely changed the way the world functions. To date, there have been over 150 million cases around the world, and counting. Similar to many respiratory viruses, the COVID-19 virus has rapidly mutated into dozens of prolific strains. The current vaccine has been designed using the original strains sequence from Wuhan, China. However, there is reason for speculation that the vaccine may not be as effective with the other dominant strains. Our project aims to provide scientists with a tool to help them compare these strains and predict characteristics that can aid in determining key factors such as vaccine efficacy. This can also be used to assist public health officials target specific strains of any virus and develop further preventative measures.
- Speed: Determine if clustering is quicker than MEGA
- Reliability: Determine if clustering results accurately group sequences
- Utilized Severe Acute Respiratory Syndrome Coronavirus 2 Data Hub to retrieve nucleotide sequnces of various strains
- Manually downloaded 228 sequences in .fasta format, ensuring 3-4 sequences from each of the 6 populated continents for each month from January 2020 - January 2021
- Parsed .fasta file, retrieving nucleotide sequences and sequence ID
- Depending on Clustering method used, trimmed sequences to a uniform length
- Fed ID and sequence into 2x2 matrix
- Converted from string to Array
- K-Means
- Defined levenshtein function and applied to sequences to produce similarity ratios
- Produced square matrix containing values
- Applied K-Means clustering algorithm on matrix using Levenshtein Distance Metric
- Agglomerative
- Formatted matrix so that each sequence is compared against reference (X) and UK Variant (Y)
- Defined levenshtein function and applied to sequences to produce similarity ratios
- Applied Agglomerative clustering on these values using L1 or Euclidean Affinity
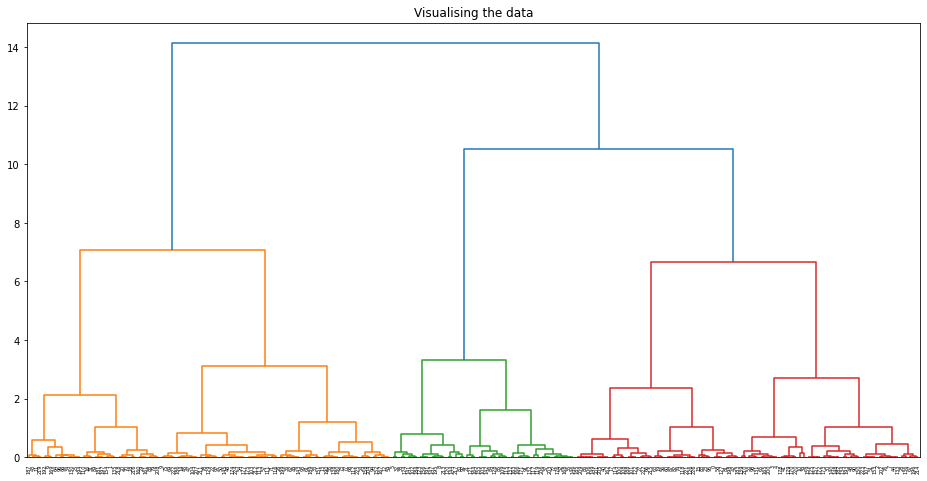
- Produced scatter plots for all clustering results
- Due to lack of uniformity in order of clustering results, produced a hash-map to align cluster labels with MEGA sample data
- Computed Accuracy and Purity scores for each result
Sklearn, plotly, Pandas, numPy, Matplot, Biopython
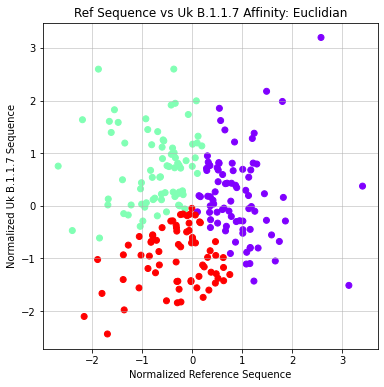
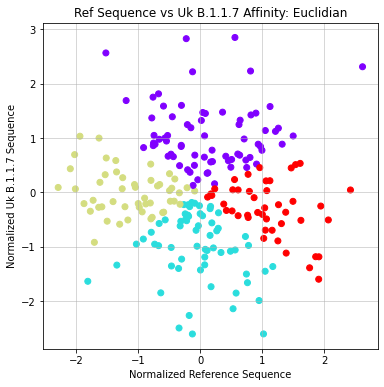
Euclidean

L1
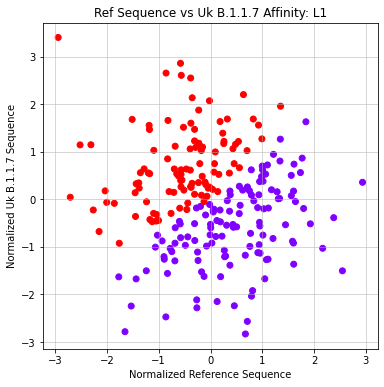

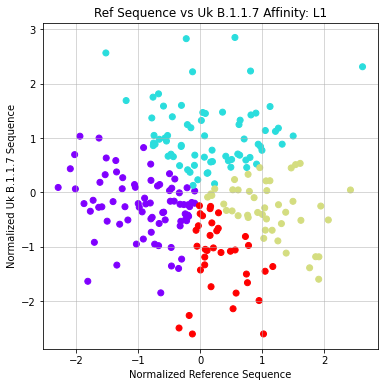
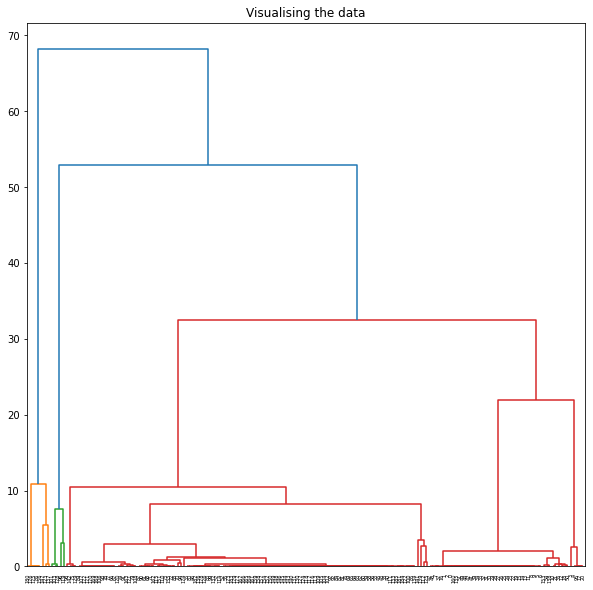
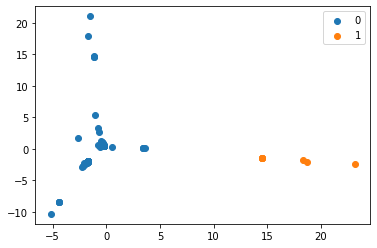
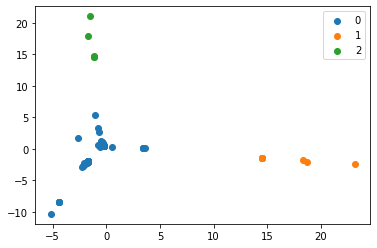
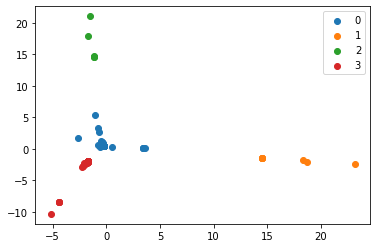

Our preliminary results show that there is promise to this methodology. Our models show that they perform better than random chance and with the proper adjustments, can be made to be highly accurate and efficient. It is also important to note that 9 different models were run simultaneously and produced results in under a minute, 4.5 times faster than MEGA. Ideally, these models can lead to efficient and correct predictions of vaccine efficacy, resistance and more on not only COVID but other fast-mutating viruses as well.
In the future, we plan on implementing:
- Streamlined data retrieval from database
- Implement gene-specific Distance Metric
- Experiment with a wider variety of clustering methods
- Divya Gollapalli
- Rushi Surampudi
- Areeba Qazi
- Shriya Jejurkar
- Team Lead - Bryant Hou
- Faculty Advisor - Dr. Anjum Chida